|
| |
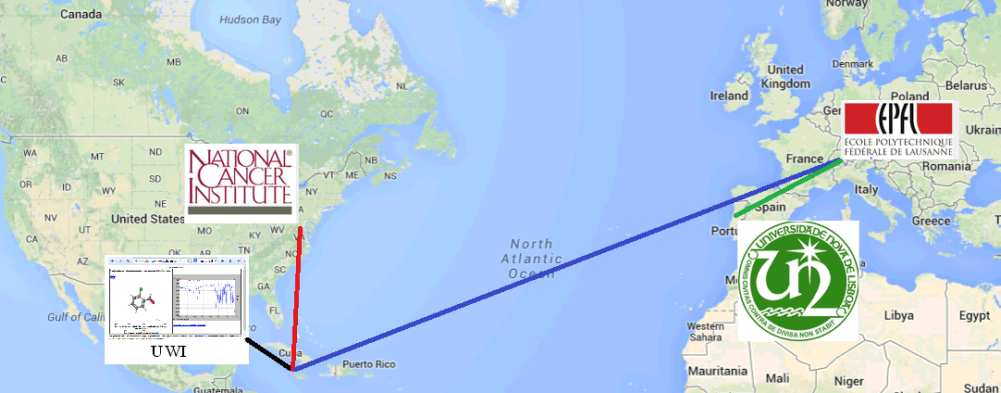
This page illustrates how we can use JSME (the JavaScript Molecular Editor)
along with JSpecView to quickly get a simulated spectrum for a compound of our choice.
Note that the three applets can be combined easily in any combination.
JSmol on this page calls servers in Frederick, Maryland (NIH resolver, for name-to-structure)
and Lausanne, Switzerland (nmrdb, for structure-to-spectrum)
Draw a chemical structure
or search for a chemical identifier such as caffeine
RYYVLZVUVIJVGH-UHFFFAOYSA-N or CCOCC,
then press .
Note that these spectra are calculated i.e. predictions.
They may differ significantly from actual NMR spectra.