Magnetism
Introduction
Movement of an electrical charge (which is the basis of electric
currents) generates a magnetic field in a material. Magnetism is
therefore a characteristic property of all materials that contain
electrically charged particles and for most purposes can be
considered to be entirely of electronic origin.
The Right Hand Rule for an induced magnetic field
In an atom, the
magnetic field is due to the coupled spin and orbital magnetic
moments associated with the motion of electrons. The spin
magnetic moment is due to the precession of the electrons about
their own axes wherease the orbital magnetic moment is due to the
motion of electrons around the nucleus. The resultant combination
of the spin and orbital magnetic moments of the constituent atoms
of a material gives rise to the observed magnetic properties.
Historically, magnetism has been recognised for thousands of
years. An account, that is probably apochryphal, tells of a
shepherd called Magnes in Crete who around 900 B.C discovered the
naturally occurring magnet lodestone (a form of the the spinel
magnetite, Fe3O4) in a region later named
Magnesia. Supposedly while he was walking over a deposit, the
lodestone pulled the nails out of his sandals and the metal tip
from his staff.
The Classical Theory of Magnetism
The classical theory of magnetism was well developed before
quantum mechanics.
Lenz's Law (~1834), states that:
when a substance is placed within a magnetic field,
H,
the field within the substance,
B, differs from H by the
induced field, 4πI,
which is proportional to the intensity of magnetization, I.
That is; B = H + 4πI
where B is the magnetic field within the
substance
H is the applied magnetic field
and I is the intensity of magnetisation
This can also be written as
B/H = 1 + 4π I/H, or
B/H = 1 + 4πκ
where B/H is called the magnetic permeability of the material and
κ is the magnetic susceptibility per unit volume, (I/H)
By definition, κ in a vacuum is zero, so under those conditions
the equation would reduce to B=H.
It is usually more convenient to measure mass than volume and the
mass susceptibility, χg, is related to the volume
susceptibility, κ, through the density.
χg = κ /ρ
where ρ is the density.
Finally to get our measured quantity on a basis that can be
related to atomic properties, we convert to molar susceptibility
χm =χg * RMM
Since this value includes the underlying diamagnetism of paired
electrons, it is necessary to correct for the diamagnetic portion
of χm to get a corrected paramagnetic
susceptibility.
χ'm = χm +
χdia
Examples of these corrections are tabulated in
the Laboratory Manual and are available on-line as well.
There are numerous methods for measuring magnetic
susceptibilites, including, the Gouy, Evans and Faraday
methods. These all depend on measuring the force exerted
upon a sample when it is placed in a magnetic field. The more
paramagnetic the sample, the more strongly it will be drawn
toward the more intense part of the field.
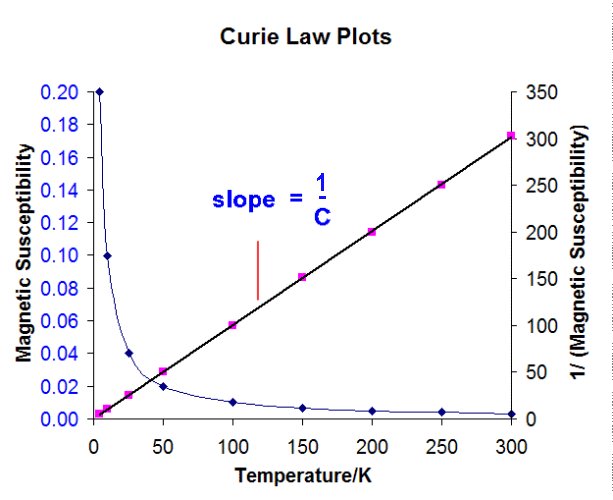
Curie Law
Normal paramagnetic substances obey the Curie Law
χ = C/T
where C is the Curie constant. Thus a plot of 1/ χ versus T
should give a straight line of slope 1/C passing through the
origin (0K).
Whereas many substances do give a straight line it often intercepts just a
little above 0K and these are said to obey the Curie-Weiss Law:
χ =C/(T+Φ)
where Φ is known as the Weiss constant.
Quantum Mechanics Approach
A similar expression (where χ is inversely proportional to Temperature)
is obtained but now the constant C is given by the
Langevin expression, which relates the susceptibility to the magnetic moment:
χm =N μ2 / 3kT
where N is Avogadro's number
k is the Boltzmann constant
and T the absolute temperature
rewriting this gives the magnetic moment as
μ= 2.828 √ {χmT} B.M.
There are two main types of magnetic compounds, those that are
diamagnetic (compounds that are repelled by a magnetic field) and
those that are paramagnetic (compounds that are attracted by a
magnetic field). All substances possess the property of
diamagnetism due to the presence of closed shells of electrons
within the substance. Note that diamagnetism is a weak effect
while paramagnetism is a much stronger effect.
Paramagnetism derives from the spin and orbital angular momenta
of electrons. This type of magnetism occurs only in compounds
containing unpaired electrons, as the spin and orbital angular
momenta is cancelled out when the electrons exist in pairs.
Compounds in which the paramagnetic centres are separated by
diamagnetic atoms within the sample are said to be magnetically
dilute.
If the diamagnetic atoms are removed from the system then the
paramagnetic centres interact with each other. This interaction
leads to ferromagnetism (in the case where the neighbouring
magnetic dipoles are aligned in the same direction) and
antiferromagnetism (where the neighbouring magnetic dipoles are
aligned in alternate directions).
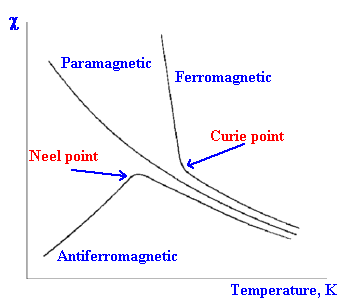
These two forms of paramagnetism show characteristic variations
of the magnetic susceptibility with temperature.
In the case of ferromagnetism, above the Curie point the material
displays "normal" paramagnetic behaviour. Below the Curie point
the material displays strong magnetic properties.
Ferromagnetism is commonly found in compounds containing iron and
in alloys.
For antiferromagnetism, above the Neel point the material
displays "normal" paramagnetic behaviour. Below the Neel point
the material displays weak magnetic properties which at lower and
lower temperatures can become essentially diamagnetic.
Antiferromagnetism is more common and is found to occur in
transition metal halides and oxides, such as TiCl3 and
VCl2.
Determination of magnetic susceptibility
The Gouy Method.
The underlying theory of the Gouy method is described
here and a form for
calculating the magnetic moment from the collected data is
available as well.
The Evans method.
The Evans balance measures the change in current required to keep
a pair of suspended magnets in place or balanced after the
interaction of the magnetic field with the sample.
The Evans balance differs from that of the Gouy in that, in the
former the permanent magnets are suspended and the position of
the sample is kept constant while in the latter the position of
the magnet is constant and the sample is suspended between the
magnets.
Orbital contribution to magnetic moments
From a quantum mechanics viewpoint, the magnetic moment is
dependent on both spin and orbital angular momentum
contributions. The spin-only formula used last year was given as:
μs.o. = √{4S(S+1)}
and this can be modified to include the orbital angular momentum
μS+L = √{4S(S+1) + L(L+1)}
An orbital angular momentum contribution is expected when the
ground term is triply degenerate i.e. a T state. These show
temperature dependence as well.
In order for an electron to contribute to the orbital angular
momentum the orbital in which it resides must be able to
transform into an exactly identical and degenerate orbital by a
simple rotation (it is the rotation of the electrons that
induces the orbital contribution). For example, in an octahedral
complex the degenerate t2g set of orbitals
(dxz,dyx,dyz) can be
interconverted by a 90o rotation. However the orbitals
in the eg subset (dz2,dx2-y2)
cannot be interconverted by rotation about any axis as the
orbital shapes are different; therefore an electron in the
eg set does not contribute to the orbital angular
momentum and is said to be quenched. In the free ion case the
electrons can be transformed between any of the orbitals as they
are all degenerate, but there will still be partial orbital
quenching as the orbitals are not identical.
Electrons in the t2g set do not always contribute to
the orbital angular moment. For example in the d3,
t2g3 case, an electron in the
dxz orbital cannot by rotation be placed in the
dyz orbital as the orbital already has an electron of
the same spin. This process is also called quenching.
Tetrahedral complexes can be treated in a similar way with the
exception that we fill the e orbitals first, and the electrons in
these do not contribute to the orbital angular momentum.
The tables in the links below give a list of all d1 to
d9 configurations including high and low spin
complexes and a statement of whether or not a direct orbital
contribution is expected.
Octahedral complexes
Tetrahedral complexes
A and E ground terms
The configurations corresponding to the A1 (free ion S
term), E (free ion D term), or A2 (from F term) do not
have a direct contribute to the orbital angular momentum.
For the A2 and E terms there is always a higher T term
of the same multiplicity as the ground term which can affect the
magnetic moment (usually by a only small amount).
μeff = μs.o. (1-α
λ /Δ)
where α is a constant (2 for an E term, 4 for an
A2 term)
λ is the spin-orbit coupling constant which is generally only available
for the free ion but this does give important information since the
sign of the value varies depending on the orbital occupancy.
some spin-orbit coupling constants for 1st row TM ions
| metal ion |
Ti(III) |
V(III) |
Cr(III) |
Mn(III) |
Fe(II) |
Co(II) |
Ni(II) |
Cu(II) |
| d configuration |
1 |
2 |
3 |
4 |
6 |
7 |
8 |
9 |
| λ / cm-1 |
155 |
105 |
90 |
88 |
-102 |
-172 |
-315 |
-830 |
for d1 to d4 the value is positive hence μeff is less than
μs.o.
for d6 to d9 the value is negative hence μeff is greater than
μs.o.
Δ is the crystal field splitting factor which again is
often not available for complexes.
For the tetrahedral Co(II) ion, CoCl42-,
the observed experimental magnetic moment, μobs = 4.59 Bohr Magneton (B.M.)
The spin-only magnetic moment, μs.o. = 3.88 B.M. which is not in good agreement.
How can we improve the analysis?
Since the ground term in the tetrahedral field is split from a 4F to
a 4A2 term then we can apply the formula above.
For an A term the constant α = 4. The spin-orbit coupling constant, λ
for the free ion is -172 cm-1 which we can use as an approximation
and Δ= 3100 cm-1.
Hence μeff = 3.88 x (1 - (4* -172) / 3100)
which comes out at μeff = 4.73 B.M.
This gives a much better fit than the spin-only formula.
In the case of the series;
CoI42-, CoBr42-, CoCl42-, Co(NCS)42-
the magnetic moments have been recorded as
4.77, 4.65, 4.59, 4.40 BM
assuming that λ is roughly a constant, then this variation
shows the inverse effect of the spectrochemical series on the magnetic moment,
since Δ is expected to increase from I- to NCS-.
T ground terms
The configurations corresponding to the T2 term (from D)
or a T1 term (from an F term) are those where there is
a direct contribution to orbital angular momentum expected.
The magnetic moments of complexes with T terms are often found to
show considerable temperature dependence. This is as a result of
spin-orbit coupling that produces levels whose energy differences
are frequently of the order kT, so as a result,
temperature will have a direct effect on the population of the
levels arising in the magnetic field.
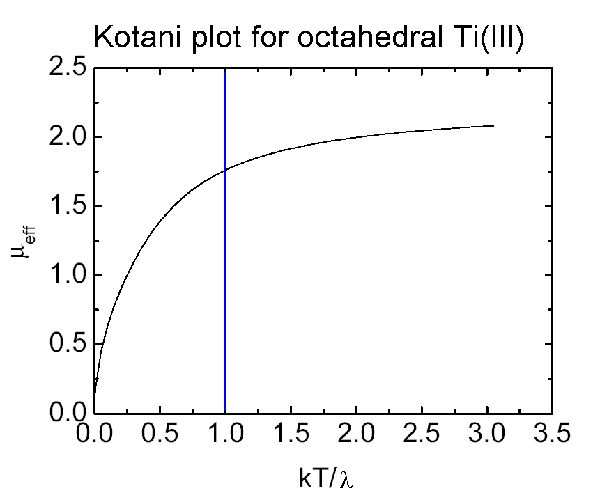
In a Kotani plot μeff is plotted against kT/λ
and when this corresponds to a value of 1 then μ equals the "spin-only"
value. If this is extrapolated to infinity then the value corresponds
to μS+L.
Measuring the magnetic moment at 80K and 300K often shows up this variation with
temperature.
A worked example.
Account for the magnetic moments of the complex,
(Et4N)2[NiCl4] recorded at 80, 99
and 300 K.
80K 99K 300K
3.25 3.43 3.89 B.M.
Ni2+ is a d8 metal ion.
The formula suggests a 4 coordinate complex and we can assume
that the complex is tetrahedral with a d electron configuration
of e4 t24 therefore the
spin-only magnetic moment can be calculated as 2.83 BM.
Why did we ignore the possibility of it being
square-planar?
The free ion Russell-Saunders ground term is 3F (L=3
and S=1) which will give rise to a lowest energy T term in a
tetrahedral field and hence the resultant magnetic moment is
expected to be temperature dependent and have a direct orbital
contribution.
The observed values may be quite different then to the calculated
spin only magnetic moment.
The value of μS+L can be calculated as:
μS+L= √{4S(S+1)+L(L+1)}
or μS+L= √{8+12}
or μS+L = √20 = 4.472B.M.
From the observed values it can be seen that the magnetic moment
of the d8 Ni2+ complex is intermediate
between the μso and μS+L values
(probably due to partial quenching of the orbital angular
momentum contribution) and is dependent on temperature.
Further worked examples and some selected magnetic data are available.
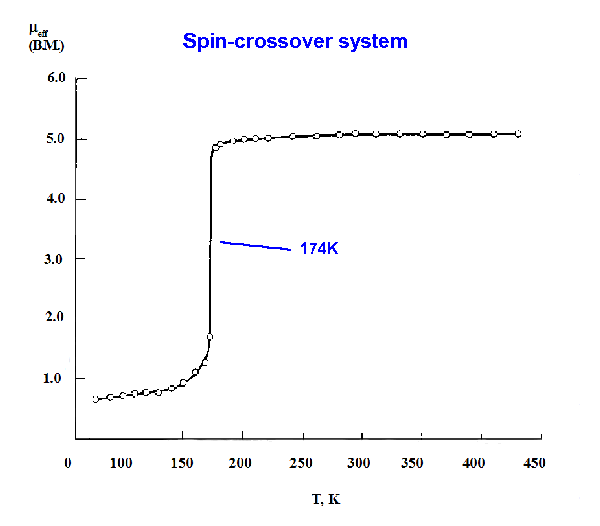
High-spin / Low-spin crossovers
Octahedral complexes with between 4 and 7 d electrons can be either high-spin
or low-spin depending on the size of Δ When the ligand field splitting
has an intermediate value such that the two states have similar energies, then
the two states can coexist in measurable amounts at equilibrium. Many "crossover"
systems of this type have been studied, particularly for iron complexes.
In the d6 case of Fe(phen)2(NCS)2, the crossover involves going from S=2 to S=0.
At the higher temperature the ground state is 5T2g while at low temperatures it
changes to 1A1g. The changeover is found at about 174K.
In solution studies, it is possible to calculate the heat of
conversion from the one isomer to the other.
return to the CHEM2101 (C21J) course outline
 Return to Chemistry, UWI-Mona,
Home Page
Return to Chemistry, UWI-Mona,
Home Page
Copyright © 2000-2011 by Robert John
Lancashire, all rights reserved.
Created and maintained by Prof. Robert J.
Lancashire,
The Department of Chemistry, University of the West Indies,
Mona Campus, Kingston 7, Jamaica.
Created July 2000. Links checked and/or last
modified 30th March 2011.
URL
http://wwwchem.uwimona.edu.jm/courses/magnetism.html